16p11.2-mikroduplikaatio-oireyhtymä
Mikroduplikaatio 16p11.2-oireyhtymä voi olla täysin oireeton tai aiheuttaa erilaisia terveysongelmia. Kromosomimutaation eli muutoksen, tässä duplikaation sijainti ja koko voi vaihdella yksilöllisesti. Sillä voi olla oma vaikutuksensa mahdolliseen oirekuvaan.
Lääketieteen toimittaja Johanna Rintahaka, Harvinaiskeskus Norio 25.9.2024
ORPHA:370079
ICD-10: Q92.3
OMIM: 614671
Avainsanat: Microduplication 16p11.2, Proximal/Distal dup(16)(p11.2), Proximal/Distal trisomy 16p11.2
Lyhyesti
Kromosomin 16p11.2-mikroduplikaatio on harvinainen kromosomimuutos. Se johtuu kromosomialueen duplikaatiosta eli kahdentumasta tietyllä alueella kromosomissa 16 (16p11.2). Usein tähän kromosomimutaatioon liittyy kehitysviive, oppimisvaikeuksia tai kehitysvamma sekä tiettyjä käyttäytymispiirteitä, kuten autismikirjon häiriö ja mahdollisuus psykiatriseen oireiluun. Lisäksi kehon rakenne voi olla hoikka ja pitkä. Osalle 16p11.2-mikroduplikaatio ei aiheuta oireita. Koska 16p11.2-mikroduplikaatiosta tiedetään vasta vähän, alla oleva diagnoosin yleisesittely on monelta osin suppea. Se ei myöskään erittele millä alueella tarkemmin 16p11.2-mikroduplikaatio on tapahtunut ja mitä geenejä on duplikaation seurauksesta tavanomaista enemmän. Mahdollisessa oirekuvassa on siis merkittävää yksilöllistä vaihtelua. Kaikkia tässä kuvattuja oireita ei siis ole kaikilla, joilla on 16p11.2-mikroduplikaatio. Vastaavasti oirekuvaan voi liittyä myös joitakin muita piirteitä, joita ei tässä kuvauksessa ole mainittu.
Oireet ja löydökset
Joillakin vastasyntyneillä 16p11.2-mikroduplikaatioon liittyy hypotonisuutta eli alentunutta lihasjänteyttä, joka voi vaikeuttaa kykyä imeä ja niellä sekä myöhemmin haitata puhumisen ja liikkumisen perustaitojen opettelua. Se voi olla osasyynä myös mahdolliseen ummetukseen. Usein hypotonia hellittää lapsen varttuessa.
Lapsella voi olla joitakin lieviä kasvonpiirteiden poikkeavuuksia, joita usein vain harjaantunut silmä osaa erottaa. Osalla todetaan mikrokefalia eli tavanomaista pienempi päänympärys. Lapsen painon kehitys voi olla heikkoa toisin kuin 16p11.2-mikrodeleetio-oireyhtymässä. Mikrodeleetio-oireyhtymässä tältä kromosomialueelta puuttuu perimäainesta. Lapsen olemus on laiha, sillä he ovat usein pitkiä suhteessa pieneen painoonsa.
Toisinaan 16p11.2-mikroduplikaatioon liittyy synnynnäisiä rakennepoikkeavuuksia, jotka voivat käsittää eri elimiä tai elinjärjestelmiä ja aiheuttaa joitakin terveyspulmia. Synnynnäisten rakennemuutosten ja 16p11.2-mikroduplikaatioon yhteys toisiinsa on kuitenkin vielä nykytiedon valossa epäselvä.
Kromosomin 16p11.2-mikroduplikaatiossa yleistä on etenkin puheen kehitysviive. Useimmiten ensisanat sanotaan 16 kuukauden ja 4–5 vuoden välisenä aikana tai myöhemmin. Vaihteluväli on siis suurta. Kaikki eivät opi puhumaan selkeästi. Niillä, joilla on puhumisen haasteita, elekielen, äänteiden tai muiden kommunikaatiotapojen merkitys korostuu. Joskus 16p11.2-mikroduplikaatioon liittyy myös puheen ymmärtämisen haasteita.
Kehitysviive voi ilmetä myös lapsen motoristen perustaitojen, kuten istumisen, liikkumisen ja kävelemisen, oppimisessa. Nämä viiveet ovat usein vähäisempiä kuin puheeseen liittyvät haasteet. Kävelemään saatetaan keskimääräisesti oppia hiukan tavanomaista myöhemmin, noin 11–18 kuukauden tietämillä. Taaperoiässä lapset saattavat kompastella, ja he voivat oppivat kiipeämään portaita tavallista myöhemmin, esimerkiksi kahden vuoden ikäisinä.
Kromosomin 16p11.2-mikroduplikaatiossa esiintyy usein oppimisvaikeuksia tai toisinaan kehitysvamma. Oppimisvaikeuksista huolimatta useimmat oppivat lukemaan, kirjoittamaan ja käyttämään tietokonetta. Oppimisen tukemisen tarve nousee selkeämmin esiin usein ylemmillä koululuokilla tai opiskeluasteilla. Mahdollisen kehitysvamman aste vaihtelee, mutta on usein lievä.
Kromosomin 16p11.2-mikroduplikaatiossa voi ilmetä ylivilkkautta ja ADHD:tä eli aktiivisuuden ja tarkkaavuuden häiriötä, levottomuutta sekä muita käyttäytymisen haasteita. Kromosomin 16p11.2-mikroduplikaatio on eräs yleisimmistä kromosomimuutoksista 16p11.2-mikrodeleetion kanssa, jotka on yhdistetty autismikirjon häiriöön. Kuitenkaan läheskään kaikilla henkilöillä ei ole autismikirjon häiriötä tai autistisia piirteitä. Mikroduplikaatio 16p11.2 voi altistaa myös psykiatriseen oireiluun, kuten kaksisuuntaiseen mielialahäiriöön ja skitsofreniaan. Nämä oireet voivat ilmaantua myöhemmällä iällä. Myös masennus ja unihäiriöt ovat mahdollisia.
Osalla ilmenee epilepsiaa. Usein ensimmäinen kohtaus ilmaantuu alle vuoden ikäisellä vauvalla. Epilepsia voi hävitä tai lievittyä lapsen vanhetessa. Epilepsia voi olla yhteydessä QPRT-, DOC2A– ja SEZ6L2-geenien kahdentumiseen. Yleensä epilepsia saadaan lääkityksellä hyvin hallintaan.
Syy ja perinnöllisyys
Kromosomin 16p11.2-mikroduplikaation oireet johtuvat kromosomialueen ja siinä sijaitsevien geenien kahdentumisesta kromosomissa 16 (16p11.2). Kahdentuneen kromosomialueen pituus ja sijainti kromosomissa vaihtelevat yksilöllisesti. Kromosomin 16p11.2-duplikaation alueella on noin 27 tunnettua geeniä. Niiden lukumäärämuutokset voivat aiheuttaa oireita. Myös oireettomuus on mahdollista.
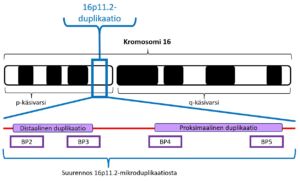
Kromosomin 16p11.2-mikroduplikaatio periytyy autosomissa dominoivasti eli vallitsevasti. Tämä tarkoittaa sitä, että vain toisessa kromosomista 16 oleva mikroduplikaatio on riittävä aikaansaamaan oireyhtymän mahdolliset oireet. Useimmiten kromosomimuutos on periytynyt lapselle jommaltakummalta vanhemmaltaan. Jos 16p11.2-mikroduplikaatio on vanhemman perimässä, kromosomimuutoksen toistumistodennäköisyys perheessä on 50 %. Tämä todennäköisyys on sama kaikissa raskauksissa. Lapsen oirekuva voi olla kuitenkin erilainen kuin hänen vanhemmallaan.
Noin neljällä kymmenestä (eli 25 %:lla) 16p11.2-mikroduplikaatio on seurausta täysin uudesta, niin kutsutusta de novo-mutaatiosta. Vanhempien perimäntutkimuksista ei tällöin löydetä 16p11.2-mikroduplikaatiota. De novo-mutaatio syntyy hedelmöitykseen osallistuvan sukusolun, munasolun tai siittiön, kypsyessä tai pian hedelmöityksen jälkeen. De novo-mutaatioiden syntyyn ei voi vaikuttaa, eivätkä ne johdu mistään asiasta, jonka vanhemmat ovat tehneet tai jättäneet tekemättä. De novo-mutaatioiden toistumistodennäköisyys perheessä on noin prosentin (1 %) eli yhden sadasosan luokkaa.
On mahdollista, joskin erittäin harvinaista, että 16p11.2-mikroduplikaatio on seurausta sukusolumosaikismista. Silloin osassa vanhemman sukusoluja on 16p11.2-mikroduplikaatio ja osassa ei. Sukusolumosaikismi nostaa jonkin verran, muttei merkittävästi, perheen todennäköisyyttä saada lapsi, jolla on 16p11.2-mikroduplikaatio. Sukusolumosaikismista lisää tietoa löytyy Harvinaiskeskus Norion sivuilta kohdasta 70 kysymystä ja vastausta perimästä ja perinnöllisyydestä: Mosaikismi.
Perhe voi halutessaan keskustella perinnöllisyysneuvonnassa oireyhtymän toistumistodennäköisyydestä jo ennen seuraavan mahdollisen raskauden alkua. Harvinaiskeskus Norion sivuilta löytyy tietoa myös perhesuunnittelusta tilanteissa, joissa perheessä on mahdollisesti kohonnut todennäköisyys johonkin harvinaissairauteen: Perhesuunnittelu ja raskaus. Kaikista näistä aiheista voi myös keskustella ilman lähetettä ja veloituksetta Harvinaiskeskus Norion perinnöllisyyshoitajan kanssa. Yhteystiedot keskustelutukeen ja neuvontaan löydät tämän julkaisun lopusta.
Yleisyys
Kromosomin 16p11.2-mikroduplikaatio yleisyydestä on useita eri arvioita lääketieteellisessä kirjallisuudessa. Todennäköisesti se on alidiagnosoitu, koska osalle kromosomimuutos ei aiheuta minkäänlaisia oireita, ja täten henkilö ei hakeudu terveydellisistä syistä perinnöllisyystutkimuksiin. Eräs arvio 16p11.2-mikroduplikaation yleisyydestä on kolme tapausta 10 000 henkilöä kohden (1: 3333). Toisen arvion mukaan proksimaalisten duplikaatioiden yleisyys on 1:2500 ja distaalisten duplikaatioiden 1:1500. Vuonna 2024 henkilöitä, joilta on tunnistettu 16p11.2-mikroduplikaatio perimästä, on yli 150. Eriteltyä tietoa 16p11.2-duplikaation yleisyydestä duplikaatiotyypin mukaan (proksimaalinen, distaalinen tai näitä suurempi) ei ole saatavilla.
Diagnoosi ja hoito
Pelkän oirekuvan perusteella diagnoosia on vaikea asettaa ja siksi geneettiset eli perimään liittyvät tutkimukset ovat välttämättömiä. Kromosomin 16p11.2-mikroduplikaatio-oireyhtymän diagnoosi voidaan asettaa, jos henkilön perimästä löydetään tällainen muutos. Usein diagnoosi helpottaa perheen tilannetta, ja voi olla merkityksellinen joissain tilanteissa myös perhesuunnittelun kannalta.
Kromosomin 16p11.2-mikroduplikaatiota ei voida korjata tai parantaa. Hoito määräytyy oireiden ja tarpeiden mukaan ja voi käsittää esimerkiksi toiminta-, puhe- ja fysioterapian. Säännöllinen oppimisen tukeminen ja kehityksen seuranta ovat tärkeitä. Mahdollisten käyttäytymiseen liittyvien pulmien mahdollisimman varhainen tunnistaminen ja kuntoutus tukee ja ylläpitää henkilön toimintakykyä. Jos epilepsiaa on, sen hoito pyrkii kohtauksettomuuteen, kohtaustiheyden harventamiseen tai kohtauksien lievittymiseen. Koko perheen huomioiminen on tärkeää ja voi ylläpitää ja lisätä perheen voimavaroja.
Ennuste
Lääketieteellisessä kirjallosuudessa ei ole mainintoja lyhentyneestä eliniästä.
Historia
Kromosomin 16p11.2-mikroduplikaatio tunnistettiin 2000-luvulla.
Kokemustieto
Kokemustietoa 16p11.2-mikroduplikaatiosta löydät Tukiliiton tarinat-osiosta Äidin ja tyttären tarina: kun sinulla on hieman enemmän kuin muilla.
SimonsSearchLight-yhteisön sivuilta löytyy useita englanninkielisiä kokemustarinoita, kohdasta ”Family Stories”.
Onko sinulla omakohtaista kokemusta tästä diagnoosista? Keräämme kokemustietotarinoita, ja sinäkin voit osallistua. Lue lisää Kokemustietoa-sivulta.
Tukipalvelut
Harvinaiskeskus Noriosta voi tiedustella vertaistukea. Lue lisää Vertaistuki-sivultamme.
Harvinaiskeskus Norion perinnöllisyyshoitajaan voi ottaa yhteyttä, kun haluaa keskustella perimään tai harvinaissairauksiin liittyvistä asioista. Lue lisää Keskustelutuki ja ohjaus -sivultamme tai soita 044 5765 439.
Tukiliiton sivuilta löytyy runsaasti tietoa erilaisista palveluista: Tuki ja neuvot.
Facebookissa on ”Ainutlaatuiset”-niminen keskusteluryhmä perheille, joita harvinaiset kromosomimuutokset koskettavat.
Facebookista löytyy hakusanalla ”16p11.2-microduplication” kaksi englannin kielistä keskusteluryhmää läheisille, joita kromosomimuutos jollakin tapaa koskettaa. Näiden nimet ovat ”16p11.2 deletion/ duplication We are family xx” ja ”16p11.2 Duplication”. Lisäksi SimonsSearchLight yhteisöllä on oma keskusteluryhmänsä, jonka nimi on ”16p11.2 Duplication: Simons Searchlight Community”. Jäseniksi ryhmiin pääsee pyytämällä niiden jäsenyyttä.
Aiheesta muualla
Socialstyrelsen: 16p11.2-duplikationssyndromet
Simons Searchlight.org: 16p11.2 Duplication ja 16p11.2 Duplication (Distal)
Simons Searchlight.org: 16p11.2 microduplication Syndrome Guidebook (2019)
RareChromo.org: 16p11.2 microduplications
Genetic and Rare Diseases Information Center (GARD): 16p11.2 duplication
MedlinePlus: 16p11.2 duplication
Lähteet
Orphanet: 16p11.2p12.2 microduplication syndrome
Orphanet: Proximal 16p11.2 microduplication syndrome
Online Mendelian Inheritance in Man (OMIM): Chromosome 16p11.2 duplication syndrome
Vos, N., Kleinendorst, L., van der Laan, L. et al. Evaluation of 100 Dutch cases with 16p11.2 deletion and duplication syndromes; from clinical manifestations towards personalized treatment options. Eur J Hum Genet (2024). DOI https://doi.org/10.1038/s41431-024-01601-2
Julkaistu ensimmäisen kerran Harvinaiskeskus Norion sivuilla 16.4.2019.