16p11.2 mikrodeleetio-oireyhtymä
Kromosomissa 16 on alue (16p11.2), joka on herkkä mutaatioille, kuten deleetioille eli häviämille ja duplikaatioille eli kahdentumille. Kromosomin proksimaalinen 16p11.2-deleetio on näistä yleisin. Tämä diagnoosikuvaus keskittyy kuvaamaan vain proksimaalista 16p11.2-deleetiota (BP4-BP5), jonka oireille on tyypillistä puheen, liikkumisen ja oppimisen vaikeudet. Kromosomin 16p11.2-deleetio voi olla myös oireeton.
Lääketieteen toimittaja Johanna Rintahaka, Harvinaiskeskus Norio 25.9.2024
ORPHA: 261197
ICD-10: Q93,5
ICD-11: LD44.G1
OMIM: 611913
Avainsanat: Proximal del(16)(p11.2), Proximal monosomy 16p11.2
Lyhyesti
Kromosomin proksimaalinen 16p11.2-deleetio (BP4-BP5; 29.6–30.2 Mb; noin 600 kb) on tyypillisin tämän kromosomialueen muutoksista eli mutaatioista. Lisäksi se on yleisin perimästä johtuva syy hermoston kehityshäiriölle ja autismikirjon häiriölle. Proksimaalisella tässä tarkoitetaan aluetta, joka on lähimpänä kromosomin sentromeeriä eli aluetta, joka erottaa kromosomin pienen (p) ja suuren (q) käsivarren toisistaan. Proksimaalisen 16p11.2-deleetion oireet vaihtelevat merkittävästi henkilöstä toiseen, ja osalla tämä mutaatio ei aiheuta lainkaan oireita. Tässä kuvattuja oireita ei siis ole kaikilla, ja vastaavasti oirekuvaan voi liittyä myös joitakin muita piirteitä, joita ei tässä diagnoosikuvauksessa ole mainittu. Tämä diagnoosikuvaus ei sisällä tietoja suuremmista tai pienemmistä kromosomialueen 16p11.2 deleetioista.
Oireet ja löydökset
Kromosomin proksimaalisen 16p11.2-deleetion oireet ja löydökset vaihtelevat merkittävästi, myös saman perheen jäsenten välillä. Vastasyntynyt voi olla hypotoninen eli hänen lihasjänteytensä on tavanomaista heikompi. Hypotonia voi vaikuttaa lapsen syömiskykyyn, motoristen eli liikunnallisten perustaitojen omaksumiseen ja puheen tuottamiseen. Hypotonisuus hellittää usein lapsen varttuessa.
Lapsella voi olla makrokefalia eli tavanomaista suurempi päänympärys. Muita lieviä erityispiirteitä kasvoissa voivat olla mm. matalalla sijaitsevat korvat, leveä otsa, pieni leuka, toisistaan kaukana sijaitsevat silmät ja matalat keskikasvot. Nämä kasvonpiirteet eivät ole spesifisiä eli tyypillisiä vain tälle kromosomimuutokselle, vaan voivat ilmetä myös muissa geneettisissä eli perimään liittyvissä oireyhtymissä.
Osalla proksimaalinen 16p11.2-deleetio aiheuttaa jonkinlaisen selkärangan epämuodostuman, kuten kyfoskolioosin tai hemivertebran. Kyfoskolioosi on rintarangan kyttyrän ja skolioosin eli sivuttaisen selkärangan vinouman yhdistelmä. Hemivertebralla tarkoitetaan puolestaan puolinikamaa, jossa toinen puoli nikamasta on jäänyt kehittymättä. Muita ulkonäköpiirteitä voivat olla selkärangan tai ristiluun seudulla oleva kuoppa (sacral dimple) ja toisen ja kolmannen varpaan synnynnäinen yhteen kasvu.
Kromosomin proksimaaliseen 16p11.2-deleetioon liittyy riski ylipainoon jo lapsuudessa. Useimmiten kouluikäiset ovat jo ylipainoisia, mutta merkittävä ylipaino voi kehittyä nopeasti myös myöhemmin. Ylensyönti (hyperfagia) voi olla osa oirekuvaa. Arvioiden mukaan noin puolet niistä lapsista ja aikuisista, joilla on proksimaalinen 16p11.2-deleetio, ovat ylipainoisia.
Kehitysviive on yleinen ja sen aste vaihtelee yksilöllisesti. Erityisesti puheen kehitysviive on yleinen, mutta myös motoristen eli liikkumisen perustaitojen omaksuminen voi tapahtua ikätasoa myöhemmin. Puheen kehityksen viive ilmenee etenkin puheilmaisussa, kuten sanastossa, puheen tuottamisessa ja artikulaatiossa. Puheen ymmärtäminen sujuu yleensä huomattavasti paremmin. Vaikka motoristen taidot, kuten istumaan, ryömimään ja kävelemään oppiminen, voivatkin kehittyä tavanomaista hitaammin, toistaiseksi kaikki ovat oppineet kävelemään. Lapsen mahdollinen hypotonisuus voi hidastaa sekä karkea että hienomotoristen taitojen omaksumista. Joillakin motorinen kehitysviive ilmenee vain esimerkiksi hienomotorisissa taidoissa.
Kognitiivisissa taidoissa eli tiedonkäsittelyyn liittyvissä kyvyissä, kuten oppimisessa, ajattelussa ja työmuistissa sekä tarkkaavuudessa, on suurta yksilöllistä vaihtelua. Toisilla oppimisvaikeuksia ei ole, kun taas toiset tarvitsevat jatkuvaa tukea. Osalla on kehitysvamma, joka on usein lievä.
Oirekirjoon voi kuulua epilepsia. Monilla epilepsia häviää varttumisen myötä. Myös unihäiriöt ovat mahdollisia.
Aivokuvantamisessa on usein nähtävissä aivojen koon kasvu ja tavanomaista paksumpi aivokurkiainen. Aivokurkiainen on rakenne, joka yhdistää vasemman ja oikean aivopuoliskon toisiinsa. Myös mm. pikkuaivoissa voi olla havaittavissa joitakin rakenteellisia muutoksia.
Proksimaalinen 16p11.2-deleetio yhdistyy usein autismikirjon häiriöön. Autismikirjon häiriölle ominaista ovat vaikeudet sosiaalisessa vuorovaikutuksessa ja kommunikaatiossa, todennäköisyys levottomuuteen, jäykkään ja toistavaan käyttäytymiseen sekä erityisiin mielenkiinnon kohteisiin. Ajatellaan, että proksimaalinen 16p11.2-deleetio lisää riskiä autismikirjon häiriöön, mutta ei itsessään aiheuttaisi sitä, koska kaikilla, joilla on todettu tämä kromosomimuutos, ei ole autismikirjon häiriötä.
Muita proksimaaliseen 16p11.2-deleetion liitettyjä käyttäytymispiirteitä ovat mm. ADHD eli tarkkaavuuden ja aktiivisuuden häiriö sekä yliaktiivisuus. Perheet luonnehtivat lapsiaan sosiaalisiksi.
Harvinaisempia proksimaalisen 16p11.2-deleetion oireita ja löydöksiä ovat psykiatriset oireet, näkö- ja kuulo-ongelmat sekä sydämen ja virtsaelimistön rakennepoikkeavuudet.
Syy ja perinnöllisyys
Lapsella on kromosomia nro 16 kaksi kappaletta. Toisen hän on perinyt äidiltään ja toisen isältään. Kromosomin proksimaalisessa 16p11.2-deleetiossa toisesta kromosomista nro 16 puuttuu perimäainesta alueelta 16p11.2. Tämä deleetio sijaitsee kromosomin 16 pienessä käsivarressa (p) (ks. kuva alla). Kun tätä aluetta tarkastellaan tarkemmin, havaitaan, että 16p11.2-mikrodeleetio käsittää kokonaisuudessaan useita alueita (BP2-BP5), joilla kromosomimutaatio useimmiten tapahtuu. Proksimaalinen 16p11.2-mikrodeleetio käsittää näistä alueista BP4-BP5:n. Ne sijaitsevat kromosomaalisessa DNA:ssa kohdassa 29.6–30.2 Mb (Mb, megaemästä) ja käsittää siis noin 600 000 emäksen (eli 600 kb) pituisen alueen DNA:n rakenteesta. DNA:n emäkset A (adeniini), C (sytosiini), T (tymiini) ja G (guaniini) ja niiden keskinäinen järjestys määrittävät DNA:n emäsjärjestyksen eli sekvenssin eri geeneineen ja geenien ulkopuolisine alueineen. Tällä proksimaalisella alueella sijaitsee ainakin 24 eri geeniä, joiden puuttuminen johtaa erilaisiin oireisiin. Lisää perustietoa DNA:n rakenteesta voit halutessasi lukea Harvinaiskeskus Norion kohdasta 70 kysymystä ja vastausta perimästä ja perinnöllisyydestä.
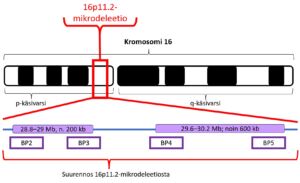
Kromosomin 16p11.2-mikrodeleetio-oireyhtymä noudattaa autosomissa dominanttia eli vallitsevaa periytymistapaa. Tämä tarkoittaa, että häviämä vain toisessa kromosomissa 16 ja sen alueella p11.2, riittää oireyhtymän kehittymiseksi. Jos vanhemmalla on tämä kromosomimuutos perimässään, hänellä on 50 %:n todennäköisyys saada lapsi, jolla on 16p11.2-mikrodeleetio. Tämä periytymistodennäköisyys on sama raskaudesta toiseen. Lapsen oirekuva voi kuitenkin olla erilainen kuin hänen vanhemmallaan. Erään arvion mukaan alle yksi lapsi kymmenestä (< 10 %) on perinyt 16p11.2-mikrodeleetion toiselta vanhemmaltaan.
Suurimmassa osassa tapauksia (n. 90 %) proksimaalinen 16p11.2-mikrodeleetio on seurausta uudesta, niin kutsutusta de novo-mutaatiosta. Vanhempien perimäntutkimuksista ei tällöin löydetä proksimaalista 16p11.2-deleetiota. De novo-mutaatio syntyy hedelmöitykseen osallistuvan sukusolun, munasolun tai siittiön, kypsyessä tai pian hedelmöityksen jälkeen. De novo-mutaatioiden syntyyn ei voi vaikuttaa, eivätkä ne johdu mistään asiasta, jonka vanhemmat ovat tehneet tai jättäneet tekemättä. De novo-mutaatioiden toistumistodennäköisyys perheessä on noin prosentin (1 %) eli yhden sadasosan luokkaa.
On mahdollista, joskin erittäin harvinaista, että oireyhtymä on seurausta sukusolumosaikismista. Silloin osassa vanhemman sukusoluja on 16p11.2-mikrodeleetio ja osassa ei. Nykyiset verikokeeseen perustuvat perimäntutkimukset eivät voi poissulkea tai vahvistaa sukusolumosaikismin mahdollisuutta. Sukusolumosaikismi nostaa jonkin verran, muttei merkittävästi, perheen todennäköisyyttä saada toinen lapsi, jolla on 16p11.2-mikrodeleetio-oireyhtymä. Sukusolumosaikismista voit halutessasi lukea Harvinaiskeskuksen sivuilta kohdasta 70 kysymystä ja vastausta perimästä ja perinnöllisyydestä: Mosaikismi.
Perhe voi halutessaan keskustella perinnöllisyysneuvonnassa oireyhtymän toistumistodennäköisyydestä jo ennen seuraavaa mahdollista raskautta. Harvinaiskeskus Norion sivuilta löytyy tietoa myös perhesuunnittelusta tilanteissa, joissa perheessä on mahdollisesti kohonnut todennäköisyys johonkin harvinaissairauteen: Perhesuunnittelu ja raskaus. Kaikista näistä aiheista voi myös keskustella ilman lähetettä ja veloituksetta Harvinaiskeskus Norion perinnöllisyyshoitajan kanssa. Yhteystiedot keskustelutukeen ja neuvontaan löydät tämän julkaisun lopusta.
Yleisyys
Kromosomin 16p11.2-mikrodeleetion todellinen yleisyys väestössä on tuntematon, koska osalla se ei aiheuta selviä oireita. Lääketieteellisessä kirjallisuudessa mainitut arviot tämän kromosomimuutoksen yleisyydestä vaihtelevat huomattavasti lähteestä toiseen. Ensimmäiset arviot olivat kolme vastasyntynyttä 10 000 lasta kohden (3:10 000). Nykyisin 16p11.2-mikrodeleetio-oireyhtymän ajatellaan olevan yleisempi: yksi vastasyntynyt 2000 lasta kohden (1:2000). Toisaalta henkilöillä, joilla on autismikirjon häiriö, 16p11.2-mikrodeleetio löydetään joka sadannelta (1:100 eli 1 %) ja yhdellä tuhannesta heistä, joilla on kielellisiä tai psykiatrisia haasteita (1:1000).
Diagnoosi ja hoito
Koska proksimaalisen 16p11.2-deleetion oirekuva vaihtelee huomattavasti, pelkän oirekuvan perusteella diagnoosia on vaikea asettaa. Tämän vuoksi geneettiset eli perimään liittyvät tutkimukset ovat välttämättömiä. Proksimaalisen 16p11.2-deleetion diagnoosi voidaan asettaa, jos perimän tutkimuksista löydetään juuri kyseinen kromosomimuutos. Epätyypillisten oireiden ja löydösten yhteydessä tarkentavat perimäntutkimukset voivat olla hyödyllisiä potilaan oirekuvan etiologian eli syyn selvittämisessä (Niels vos, 2024).
Kromosomin 16p11.2-deleetiota ei voida korjata tai parantaa, mutta sen mahdollisesti aiheuttamia oireita pystytään hoitamaan. Hoito perustuu yksilön tarpeisiin. Lapsen ja nuoren kasvua ja kehitystä seurataan säännöllisesti. Luontaista ruokahalua ja syömiskäyttäytymistä on hyvä seurata. Mahdolliseen ylipainon kertymiseen on hyvä puuttua mahdollisimman varhaisessa vaiheessa. Painonhallinnassa auttaa terveellinen ruokavalio yhdistettynä riittävään liikuntaan ja terveyttä edistävän syömiskäyttäytymisen tukemiseen. Fysio-, puhe- ja toimintaterapian tarve on yksilöllinen. Oppimisen tuen tarve vaihtelee, ja osalla jatkuva tuki on tarpeen. Autismikirjon häiriön varhainen tunnistaminen ja kuntoutus tukee ja ylläpitää henkilön toimintakykyä. Mahdollisen epilepsian hoito pyrkii kohtauksettomuuteen, kohtaustiheyden harventamiseen tai kohtauksien lievittymiseen. Koko perheen tukeminen on tärkeää.
Ennuste
Eliniän odote riippuu 16p11.2-deleetion aiheuttamista mahdollisista oireista. Toistaiseksi luotettavia arvioita elinennusteesta ei ole antaa.
Historia
Kromosomin 16p11.2-deleetio tunnistettiin ensimmäisen kerran 2000-luvun alussa.
Huomioitavaa
Distaalisesta kromosomin 16p11.2-deleetiosta (BP2-BP3, 28.8–29 Mb, noin 200 kb) tiedetään toistaiseksi vähemmän kuin proksimaalisesta deleetiosta (BP4-BP5; 29.6–30.2 Mb; noin 600 kb). Se käsittää kuitenkin osittain samoja löydöksiä ja oireita kuin proksimaalinen 16p11.2-deleetio. Myös suuremmat deleetiot ja duplikaatiot ovat mahdollisia kromosomin 16p11.2 alueella.
Kokemustietoa
Onko sinulla omakohtaista kokemusta tästä diagnoosista? Keräämme kokemustietotarinoita, ja sinäkin voit osallistua. Lue lisää Kokemustietoa-sivulta.
Tukipalvelut
Harvinaiskeskus Noriosta voi tiedustella vertaistukea. Lue lisää Vertaistuki-sivultamme.
Harvinaiskeskus Norion perinnöllisyyshoitajaan voi ottaa yhteyttä, kun haluaa keskustella perimään tai harvinaissairauksiin liittyvistä asioista. Lue lisää Keskustelutuki ja neuvonta -sivultamme tai soita 044 5765 439.
Tukiliiton sivuilta löytyy runsaasti tietoa erilaisista palveluista: Tuki ja neuvot.
Facebookista löytyy suomenkielinen keskusteluryhmä ”16p11.2 Microdeletion Finland” sekä ”Ainutlaatuiset”-niminen keskusteluryhmä perheille, joita harvinaiset kromosomimuutokset koskettavat. Lisäksi Facebookissa on useita englanninkielisiä keskusteluryhmiä. Jäseniksi FB-keskusteluryhmiin pääsee pyytämällä niiden jäsenyyttä.
Aiheesta muualla
Socialstyrelsen: 16p11.2-deletionsyndromet
Genetic and Rare Diseases Information Center (GARD): Proximal 16p11.2 microdeletion syndrome
MedlinePlus: 16p11.2-deletion syndrome
Lähteet
Orphanet: Proximal 16p11.2 microdeletion syndrome
Online Mendelian Inheritance in Man (OMIM): Chromosome 16p11.2 deletion syndrome, 593 kB
GeneReviews®: 16p11.2 Recurrent Deletion
Vos, N., Kleinendorst, L., van der Laan, L. et al. Evaluation of 100 Dutch cases with 16p11.2 deletion and duplication syndromes; from clinical manifestations towards personalized treatment options. Eur J Hum Genet (2024). DOI https://doi.org/10.1038/s41431-024-01601-2
Chung, W. K., Herrera, F. F., & Simon’s Searchlight Foundation (2024). Health supervision for children and adolescents with 16p11.2 deletion syndrome. Cold Spring Harbor molecular case studies, 9(4), a006316. https://doi.org/10.1101/mcs.a006316
Julkaistu ensimmäisen kerran Harvinaiskeskus Norion sivuilla 25.9.2024